
Polycystic kidney disease
Polikistik böbrek hastalığı (PKD veya PCKD, polikistik böbrek sendromu olarak da bilinir), böbrek tübüllerinin yapısal olarak anormal hale geldiği ve böbrekte çok sayıda kistin gelişmesine ve büyümesine neden olan genetik bir bozukluktur. [5] Bu kistler uteroda, bebeklik döneminde, çocuklukta veya yetişkinlikte gelişmeye başlayabilir. [6] Kistler, içlerine pompalanan sıvıyla dolu, işlevsiz tübüllerdir; boyut olarak mikroskobik boyuttan devasa boyutlara kadar değişir, bitişik normal tübülleri ezer ve sonunda onları işlevsiz hale getirir.
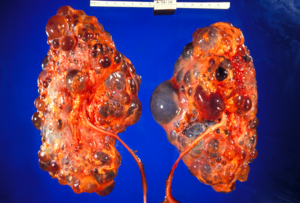
Transplantasyon sırasında çıkarılan ciddi şekilde etkilenmiş polikistik böbrekler
PKD'ye, belirli bir anormal protein üreten anormal genler neden olur; bu proteinin tübül gelişimi üzerinde olumsuz bir etkisi vardır. PKD, her biri kendi patolojisine ve genetik nedenine sahip iki tür için genel bir terimdir: otozomal dominant polikistik böbrek hastalığı (ODPKD) ve otozomal resesif polikistik böbrek hastalığı (ARPKD). Anormal gen vücuttaki tüm hücrelerde bulunur; sonuç olarak karaciğerde, seminal veziküllerde ve pankreasta kistler oluşabilir. Bu genetik kusur aynı zamanda aort kökü anevrizmalarına ve Willis serebral arterlerinin çemberinde anevrizmalara neden olabilir ve bunlar yırtılırsa subaraknoid kanamaya neden olabilir.
Teşhis şunlardan biri, bazıları veya tümünden şüphelenilebilir: yeni başlayan yan ağrısı veya kırmızı idrar; olumlu bir aile öyküsü; fizik muayenede genişlemiş böbreklerin palpasyonu; abdominal sonogramda tesadüfi bir bulgu; veya rutin laboratuar çalışmasında (BUN, serum kreatinin veya eGFR) anormal böbrek fonksiyonunun tesadüfi bir bulgusu. Kesin tanı abdominal BT incelemesi ile yapılır.
Komplikasyonlar arasında renin-anjiyotensin-aldosteron sisteminin (RAAS) aktivasyonuna bağlı hipertansiyon, sık kist enfeksiyonları, idrar kanaması ve azalan böbrek fonksiyonu bulunur. Hipertansiyon, anjiyotensin dönüştürücü enzim inhibitörleri (ACEI'ler) veya anjiyotensin reseptör blokerleri (ARB'ler) ile tedavi edilir. Enfeksiyonlar antibiyotiklerle tedavi edilir. Azalan böbrek fonksiyonu, renal replasman tedavisi (RRT) ile tedavi edilir: diyaliz ve / veya transplantasyon. Şüpheli veya kesin tanı anından itibaren yönetim, kurul onaylı bir nefrolog tarafından yapılır.
● Belirti ve bulgular
Belirti ve semptomlar arasında yüksek tansiyon, baş ağrısı, karın ağrısı, idrarda kan ve aşırı idrara çıkma yer alır. [1] Diğer semptomlar sırtta ağrı ve kist oluşumunu (böbrek ve diğer organlar) içerir. [7]
● Neden
PKD'ye, tübül gelişimi üzerinde olumsuz bir etkiye sahip olan spesifik bir anormal protein üreten anormal genler neden olur. PKD, her biri kendi patolojisi ve genetik nedeni olan iki tip için genel bir terimdir: otozomal dominant polikistik böbrek hastalığı (ODPKD) ve otozomal resesif polikistik böbrek hastalığı (ARPKD). [8] [9]
✔Otozomal dominant
Ana madde: Otozomal dominant polikistik böbrek hastalığı
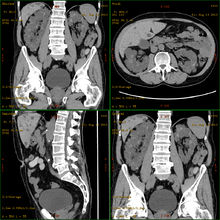
Otozomal dominant polikistik böbrek hastalığını gösteren BT taraması
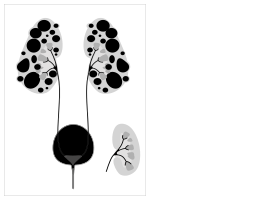
Otozomal dominant polikistik böbrek hastalığının karikatürü, diyagramın sağında normal böbrek sokması ile
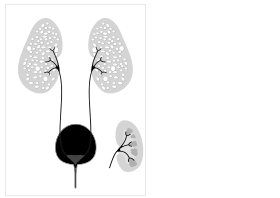
Otozomal resesif polikistik böbrek hastalığı karikatürü, diyagramın sağında normal böbrek iç içe
Otozomal dominant polikistik böbrek hastalığı (ODPBH), tüm kalıtsal kistik böbrek hastalıkları arasında en yaygın olanıdır [10] [11] [12], 1: 500 canlı doğum insidansı ile [10] [12] Çalışmalar, Avrupa ve ABD'de diyaliz ile tedavi edilen son dönem böbrek hastalığı (ESKD) hastalarının% 10'unun başlangıçta ODPBH tanısı aldığını ve tedavi edildiğini göstermektedir. [10] [9]
Üç gen PKD1, PKD2 ve PKD3'ün herhangi birindeki genetik mutasyonlar benzer fenotipik sunumlara sahiptir.
1️⃣ Gen PKD1, kromozom 16'da bulunur ve epitel hücrelerinde hücre döngüsünün ve hücre içi kalsiyum taşınmasının düzenlenmesinde rol oynayan bir proteini kodlar ve ADPBH vakalarının% 85'inden sorumludur. [13]
2️⃣ K> Na >> Ca için içe doğru seçiciliği ve Ca2 + ≈ Ba2 +> Na + ≈ K + için dışa doğru seçiciliği olan bir grup voltaj bağlantılı katyon kanalı, kromozom 4'te PKD2 tarafından kodlanmıştır [kaynak belirtilmeli]
3️⃣ PKD3 son zamanlarda araştırma makalelerinde varsayılan üçüncü bir gen olarak göründü. [10] [11] ODPBH vakalarının% 10'undan daha azı ODPBH olmayan ailelerde görülmektedir. Kist oluşumu, nefron boyunca herhangi bir noktadan uteroda başlar, ancak nefronların% 5'inden daha azının dahil olduğu düşünülmektedir. Kistler sıvı biriktirdikçe büyür, nefrondan tamamen ayrılır, komşu böbrek parankimine baskı yapar ve giderek böbrek fonksiyonunu bozar. [9]
✔ Otozomal resesif
Otozomal resesif polikistik böbrek hastalığı (ARPKD) (OMIM # 263200), 1: 20.000 canlı doğum insidansı ile iki tür PKD arasında daha az yaygındır ve tipik olarak doğumdan sonraki ilk birkaç hafta içinde tanımlanır. Ne yazık ki, böbrekler genellikle gelişmemiş durumdadır ve ARPKD'li yenidoğanlarda% 30'luk bir ölüm oranıyla sonuçlanır. PKHD1 işin içinde. [10] [9]
● Mekanizma
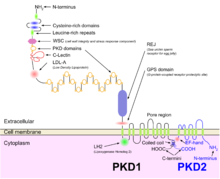
PKD1 ve PKD2
Hem otozomal dominant hem de otozomal resesif polikistik böbrek hastalığı kisti oluşumu, anormal kirpik aracılı sinyalleşmeye bağlıdır. Polikistin-1 ve polikistin-2 proteinleri, her iki proteindeki kusurlara bağlı olarak hem otozomal dominant hem de resesif polikistik böbrek hastalığına karışmış gibi görünmektedir. [14] Her iki proteinin de kalsiyum kanal proteinleri ile iletişimi vardır ve dinlenme (hücre içi) kalsiyumda ve kalsiyumun endoplazmik retikulum depolanmasında azalmaya neden olur. [15]
Hastalık, mutasyona uğramış bir dominant allelin bir ebeveynden miras alındığı ve sadece normal, vahşi tip genden sonra meydana gelen kist oluşumunun, renal tübüler ile sonuçlanan ikinci bir genetik 'vuruş' sürdürdüğü 'ikinci hit' fenomeni ile karakterize edilir. kist oluşumu ve hastalığın ilerlemesi. [16]
PKD, vücuttaki çoğu hücrenin yüzeyinde bulunan ve bazal gövde tarafından hücre gövdesine tutturulmuş, hareketsiz, kıl benzeri bir hücresel organel olan primer siliyumdaki kusurlardan kaynaklanır. [16] Böbrekte, böbrek epitelinin apikal yüzeyinden tübül lümenine çıkıntı yapan nefronun çoğu hücresinde birincil kirpikler bulunduğu bulunmuştur. Tüylerin idrar akışında büküldüğüne ve sinyallemede değişikliklere yol açtığına inanılıyordu, ancak bunun deneysel bir hata olduğu gösterildi (kirpiklerin bükülmesi, fokal düzlem telafisinin bir artefaktıydı ve ayrıca şiddetli miktürisyon üzerindeki gerçek etkiydi. hipertansiyon ve kalp durması) ve siliyanın bükülmesi Ca akışındaki değişikliklere katkıda bulunmaz. Birincil siliyumdaki kusurların kist gelişimine nasıl yol açtığı bilinmemekle birlikte, hücre içi kalsiyum, Wnt / β-katenin, siklik adenozin dahil olmak üzere birincil siliyum tarafından düzenlenen birçok sinyal yolundan birinin bozulmasıyla ilişkili olabileceği düşünülmektedir. monofosfat (cAMP) veya düzlemsel hücre polaritesi (PCP). Primer siliyumun işlevi bozulur ve kistik epitel farklılaşması, artmış hücre bölünmesi, artmış apoptoz ve resorptif kapasite kaybına neden olan bir dizi hücre içi sinyal kaskadının bozulmasına neden olur. [9] [16]
● Teşhis
Polikistik böbrek hastalığı, karın BT taramasının yanı sıra bir MRI ve aynı bölgenin ultrasonu ile tespit edilebilir. Fiziksel bir muayene / test genişlemiş karaciğer, kalp hırıltısı ve yüksek kan basıncını ortaya çıkarabilir [1]
✅ Doğal tarih
Çoğu vaka yetişkinlikte iki taraflı hastalığa ilerler. [10]
● Tedavi

Chr 11 CGAP'tan FISH ile eşlenmiş BAC'ler
FDA onaylı bir tedavi yoktur. Bununla birlikte, son araştırmalar farelerde hafif ila orta dereceli diyet kısıtlamalarının otozomal dominant polikistik böbrek hastalığının (ODPBH) ilerlemesini yavaşlattığını göstermektedir. [17]
Belirli bir vakada hastalık yeterince ilerlediğinde, nefrolog veya diğer pratisyen hekim ve hasta, son dönem böbrek hastalığını tedavi etmek için hangi tür renal replasman tedavisinin kullanılacağına karar vermelidir (böbrek yetmezliği, tipik olarak evre 4 veya 5. kronik böbrek hastalığı). [18]
Bu, değişen sıklık ve sürelerde en az iki farklı şekilde yapılabilen bir tür diyaliz olacaktır (ister evde ister klinikte, kullanılan yönteme ve hastanın stabilitesine ve eğitimine bağlıdır) ve sonunda, eğer durumlarının doğası ve ciddiyeti nedeniyle ve uygun bir eşleşme bulunursa, tek veya iki taraflı böbrek nakli için uygundurlar. [18]
Otozomal dominant polikistik böbrek hastalığı üzerine yapılan bir Cochrane Review çalışması, antibiyotik direncinden kaçınırken, böbreklerdeki kistlerin enfeksiyonlarını kontrol etmenin ve belirli bir süre için gerekli olduğunda karaciğerdeki enfeksiyonları kontrol etmenin her zaman önemli olduğuna dikkat çekti. "Bakteriyostatik ve bakteriyosidal ilaçlar" kullanarak enfeksiyonla mücadele süresi. [9] [18]
☑ Prognoz
ODPBH bireylerin normal bir yaşamı olabilir; tersine, ARPKD böbrek fonksiyon bozukluğuna neden olabilir ve 40-60 yaşlarında böbrek yetmezliğine yol açabilir. ADPKD1 ve ADPKD2 çok farklıdır, çünkü ADPKD2 çok daha hafiftir. [19]
Şu anda ODPBH'nin ilerlemesini önlemede etkili olduğu kanıtlanmış hiçbir tedavi yoktur. [20]
● Epidemiyoloji
PKD, Amerika Birleşik Devletleri'nde 600.000'den fazla insanı etkileyen en yaygın kalıtsal hastalıklardan biridir. Tüm son dönem böbrek hastalıklarının yaklaşık% 10'unun sebebidir. Erkekleri, kadınları ve tüm ırkları eşit derecede etkiler. [21] PKD, insanlarda olduğu gibi bazı hayvanlarda da görülür. [22] [23]
Kaynak : https://en.m.wikipedia.org/wiki/Polycystic_kidney_disease