Pfeiffer Sendromu
+Pfeiffer sendromu, kafatasının belirli kemiklerinin erken füzyonu (kraniyosinostoz) ile karakterize, başın ve yüzün şeklini etkileyen nadir bir genetik bozukluktur. Ek olarak, sendrom ellerde (geniş ve eğik baş parmaklar gibi) ve ayaklarda (geniş ve eğik ayak baş parmakları gibi) anormallikleri içerir. Pfeiffer sendromuna fibroblast büyüme faktörü reseptörleri FGFR1 ve FGFR2'deki mutasyonlar neden olur.
+Sendrom üç tipe ayrılmıştır; tip 1 (klasik Pfeiffer sendromu) daha hafiftir ve her iki gendeki mutasyonlardan kaynaklanır ve tip 2 ve 3 daha şiddetli olup, genellikle bebeklik döneminde FGFR2'deki mutasyonların neden olduğu ölüme yol açar. Sendromun tedavisi yok. Tedavi destekleyicidir ve genellikle hayatın ilk yıllarında kafatası deformitelerini ve solunum fonksiyonunu düzeltmek için ameliyatı içerir. Pfeiffer sendromu tip 1 olan çoğu birey normal bir zeka ve yaşam süresine sahipken, tip 2 ve 3 tipik olarak nörogelişimsel bozukluklar ve erken ölümle sonuçlanır. Pfeiffer sendromu 100.000 doğumda yaklaşık 1'i etkiler. Sendrom, adını 1964'te tanımlayan Alman genetikçi Rudolf Arthur Pfeiffer'den (1931–2012) almıştır.
+Karakteristik yüz özelliklerinin çoğu, kafatası kemiklerinin (kraniyosinostoz) erken füzyonundan kaynaklanır. Baş normal olarak büyüyemez, bu da yüksek bir alnına (turribrachycephaly) ve şişkin görünen (proptozis) ve geniş yapılı (hipertelorizm) gözlere yol açar. Ek olarak, az gelişmiş bir üst çene (maksiller hipoplazi) vardır. Pfeiffer sendromlu çocukların yüzde 50'den fazlasında işitme kaybı vardır; diş problemleri de yaygındır. Pfeiffer sendromlu kişilerde, başparmaklar ve ilk (büyük) ayak parmakları geniştir ve diğer parmaklardan (pollex varus ve hallux varus) uzağa doğru bükülür. Alışılmadık derecede kısa el ve ayak parmakları (brakidaktili) da yaygındır ve parmaklar arasında (sindaktili olarak) bir miktar ağ veya füzyon olabilir.
+Pfeiffer sendromunun en yaygın kabul gören klinik sınıflandırması 1993 yılında M. Michael Cohen tarafından yayınlandı. Cohen sendromu, tümü geniş baş parmaklar, geniş ayak parmakları, brakidaktili ve muhtemelen sindaktili olarak karakterize edilen, muhtemelen örtüşen üç türe ayırdı: Klasik Pfeiffer sendromu olarak da bilinen Tip 1, kraniyosinostoz ve "orta yüz yetmezliği" ni içerir. Bu tip, otozomal dominant bir modelde kalıtılır. Tip 1 Pfeiffer sendromlu çoğu birey normal zekaya ve normal bir yaşam süresine sahiptir. Tip 2, şiddetli proptozun yanı sıra geniş kemik füzyonu nedeniyle yonca yaprağı şeklinde bir kafatası içerir. Bu tip düzensiz olarak ortaya çıkar (yani kalıtsal görünmez) ve "kötü prognoza ve genellikle erken ölümle birlikte ciddi nörolojik uzlaşmaya" sahiptir. Tip 3, kraniyosinostoz ve şiddetli proptozu içerir. Bu tip düzensiz olarak ortaya çıkar (yani kalıtsal görünmez) ve "kötü prognoza ve genellikle erken ölümle birlikte ciddi nörolojik uzlaşmaya" sahiptir.
KAYNAK: https://en.wikipedia.org/wiki/Pfeiffer_syndrome
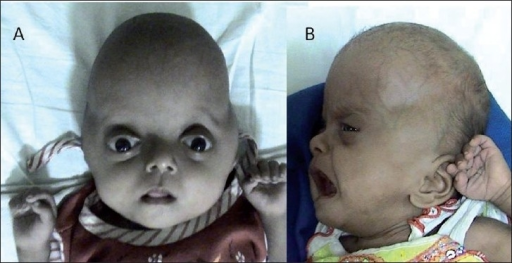 type2
type2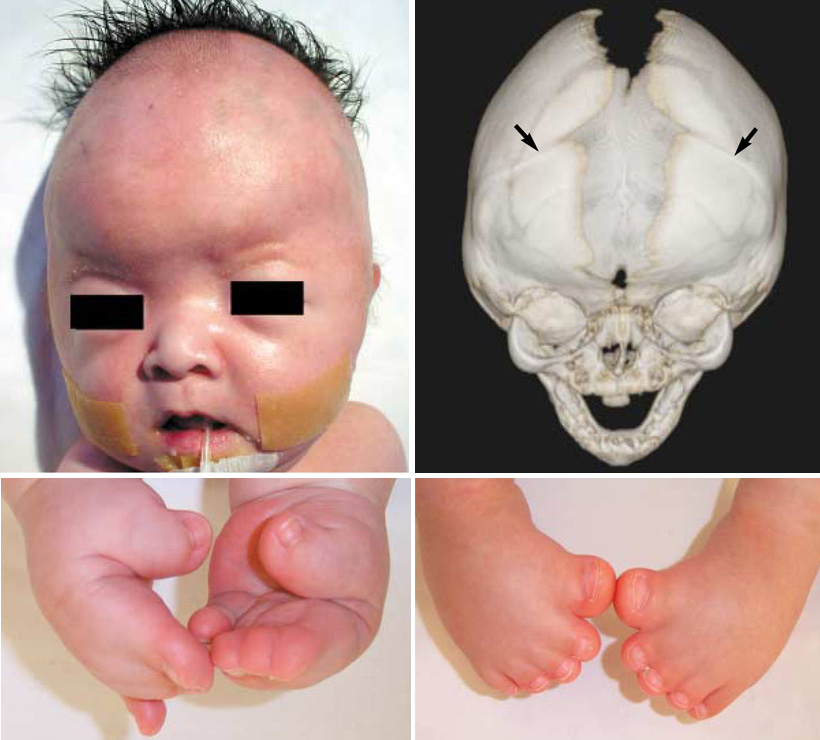 type1
type1