Crouzon Sendromu
+Crouzon sendromu, branşiyal ark sendromu olarak bilinen otozomal dominant bir genetik bozukluktur. Spesifik olarak, bu sendrom, maksilla ve mandibulanın öncüsü olan ilk branşiyal (veya faringeal) kemeri etkiler. Dallanma kemerleri büyüyen bir embriyoda önemli gelişim özellikleri olduğundan, gelişimlerindeki bozukluklar kalıcı ve yaygın etkiler yaratır.
+Bu sendrom adını, bu hastalığı ilk tanımlayan Fransız doktor Octave Crouzon'dan almıştır. İlk olarak "kraniyofasiyal disostoz" olarak adlandırılan ("kraniofasiyal", kafatası ve yüz anlamına gelir ve "disostoz", kemiğin malformasyonunu ifade eder), bu bozukluk, eski adının temel anlamlarıyla tanımlanabilen bir dizi klinik özellik ile karakterize edildi.
+Bu sendrom, 10. kromozomda bulunan fibroblast büyüme faktörü reseptörü 2'deki (FGFR2) bir mutasyondan kaynaklanır. Gelişmekte olan fetüsün kafatası ve yüz kemikleri erken kaynaşır veya genişleyemez. Böylece normal kemik büyümesi gerçekleşemez. Farklı sütürlerin füzyonu, kafatasının anormal büyüme modellerine yol açar.
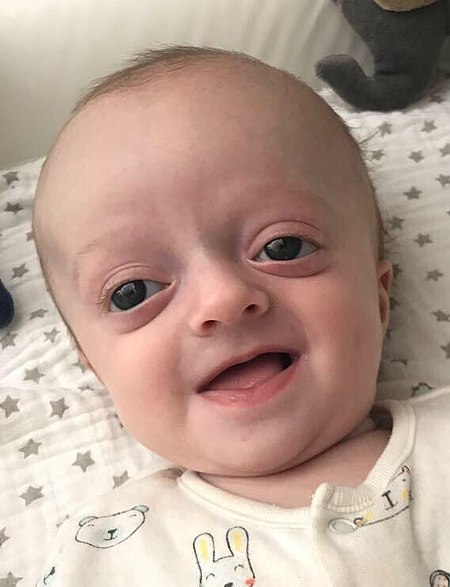
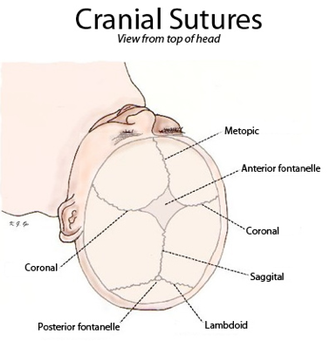
+Crouzon sendromunun tanımlayıcı bir özelliği, anormal bir kafa şekli ile sonuçlanan kraniyosinostozdur. Bu, şu kombinasyonlarda mevcuttur: turrisefali, frontal çıkıntı, trigonosefali (metopik sütürün füzyonu), brakisefali (koronal sütürün füzyonu), dolikosefali (sagittal sütürün füzyonu), plajiyosefali (lambdoid'in tek taraflı erken kapanması) , oksikefali (koronal ve lambdoidal sütürlerin füzyonu) ve karmaşık kraniyosinostoz (dikişlerin bazılarının veya tamamının erken kapanması).
+Ekzoftalmi (çevreleyen kemiklerin erken füzyonundan sonra sığ göz çukurlarından kaynaklanan şişkin gözler), hipertelorizm (gözler arasındaki normal mesafeden daha büyük) ve psittichorhina (gaga benzeri burun) da çok yaygın özelliklerdir. Pek çok vakada mevcut olan diğer yüz özellikleri arasında dış şaşılık ve hipoplastik maksilla (orta yüzün yetersiz büyümesi) yer alır, bu da göreceli mandibular prognatizm (çene çıkıntısı) ile sonuçlanır ve içbükey bir yüze sahip hastanın etkisini verir.
+Semptomların çoğu, anormal kafatası yapısına ikincildir. Crouzon sendromlu kişilerin yaklaşık% 30'u hidrosefali geliştirir. Bazı durumlarda sensörinöral işitme kaybı mevcuttur. Gözlerin göz yuvalarına sığma şeklindeki anormallikler görme sorunlarına neden olabilir, bunların en yaygın olanı görme bozukluğuna yol açabilen kornea maruziyetidir. Durumu olan bazı kişilerde hava yolu kısıtlıdır ve ciddi solunum problemleri yaşayabilir.
+Sık görülen özellikler arasında dar / yüksek kemerli damak, arka iki taraflı çapraz kapanış, hipodonti (bazı dişlerin eksik olması) ve dişlerde çapraşıklık vardır. Maksiller hipoplaziye bağlı olarak, Crouzon sendromlu kişilerde genellikle önemli ölçüde kalıcı alt kapanış vardır.
+Mevcut araştırma, otozomal dominant Crouzon sendromuna neden olan başlıca faktörlerin fibroblast büyüme faktörü reseptörleri (FGFR) FGFR2 ve FGFR3 olduğunu göstermektedir. Bu iki transmembran proteini, embriyonik gelişim sırasında osteoblast farklılaşmasında rol oynayan dört fibroblast büyüme faktörü reseptöründen ikisidir; bu reseptörler arasındaki mutasyonlar, çeşitli genetik bozukluklarda rol oynar. Çoğu yanlış anlam mutasyonundan kaynaklanan 40 bilinen mutasyon vardır. FGFR2, en yaygın mutasyona uğramış gendir, ekson 9'daki sistein 342'de bir işlev kazancı yaratan bir yanlış anlamdır. FGFR2 geninin ekson 3'ün alternatif eklenmesi yoluyla oluşturulan FGFR2lllc izoformu, ekson 9'u kullanır ve kemikleşmeyi kontrol etmek için mezenkimal kök hücrelerde kullanılır. Bununla birlikte, mutasyon, sistein 342 kaybına bağlı olarak yanlış oluşturulan bir disülfid bağı yoluyla transmembran proteinini yapısal olarak aktive eder. FGFR3, embriyonik gelişim sırasında frontal kemiklerde daha fazla ifade edilir ve kraniyal kemik gelişimine rehberlik eder. Bir nokta mutasyonu, proteinin sitosolik bölgesinde yer alan aktivasyon döngüsünde tirozinin yapısal aktivasyonuna neden olur ve frontal osteoblastların hızlı farklılaşmasına yol açar, bu da frontal kraniyal kemiklerin erken füzyonuna yol açar.
+Crouzon sendromunun teşhisi genellikle bebeğin fiziksel görünümü değerlendirilerek doğumda ortaya çıkabilir. Crouzon sendromunun teşhisini doğrulamak için radyografiler, manyetik rezonans görüntüleme (MRI) taramaları, genetik testler, X ışınları ve BT taramaları dahil olmak üzere daha fazla analiz kullanılabilir.
KAYNAK: https://en.wikipedia.org/wiki/Crouzon_syndrome