
Huntington Hastalığı - Huntingtin proteini (Htt)
20-50 yaşları arasında kendini gösteren, Iotozomal dominant geçişli bir hareket bozukluğu
Neden
- T-15 genindeki mutasyon (gen mutasyonu ile ilgili bilgilerimiz, patogenezdeki mekanizmanın anlaşılmasına yetmemektedir)
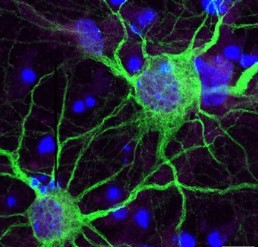
- Hastalığın özgün bileşenlerinden biri olan “huntingtin proteini (Htt)”, bu tabloya neden olan “polyQ” grubu glutamilerini içerir;
- Mutasyona uğramış olan Huntingtin proteini (Htt), nöronların sitoplazmalarında intrasellüler inklüzyon cisimleri olarak birikirler
- Corpus striatum, serebral korteks, serebellum ve medulla spinalis inklüzyon cisimlerinin en çok görüldüğü bileşenler
- İmmunohistokimyasal incelemelerde “Htt pozitif” ve “ubiquitin pozitif” nitelik gösterir
Bulgular
- İlk bulgular 35-45 yaşlar arasında
- Parkinsonizme dönüşebilen kore
- Sarsıntılı, hiperkinetik, distonik hareketler
- Motor semptomlar bilişsel bozulmadan ve kişilik değişikliklerinden önce
- Demans
- Progresif tablo (15 yıl içinde ölüm)
- Morfolojik incelemeler
- Corpus striatum'un (nucleus caudatus, putamen, globus pallidus) yoğun biçimde etkilenir
- Makroskopik incelemelerde, nucleus caudatus, nucleus accumbens, putamen ve globus pallidus atrofisi
- Nöron yitirilmesi ve gliozis mikroskopik bulguların başında gelir (nöron kayıpları, özellikle nörotransmitterler olarak γ-aminobütirik asit kullanan dikenli striatal nöronlarda saptanır)
- Corpus striatum’daki nöron kaybının düzeyi, hastalığın ileri evrelerinin göstergesi
- Son evreye doğru, serebral korteks nöronlarındaki aşırı kayıp ve buna bağlı atrofi (bu evrede, beyin korteksi nöronlarındaki Htt inklüzyonları oldukça yoğun)
13
7
0
0
0
0
0